The Shen lab is a part of the Institute of Insect Sciences and the Centre for Evolutionary & Organismal Biology at Zhejiang University. We believe that genome sequences, which are often used to understand the biodiversity that is essential to all aspects for the study of evolution and biology in general, are rich archives of history and function. Research in the lab focuses on utilizing computational approaches and genome-scale data to shed light on questions in molecular phylogenetics, evolutionary biology, comparative genomics, and bioinformatics.
Lab News
◆25/5/2026 - Please check out our cover article in Cell Research on exploring darkness in the insect tree of life!

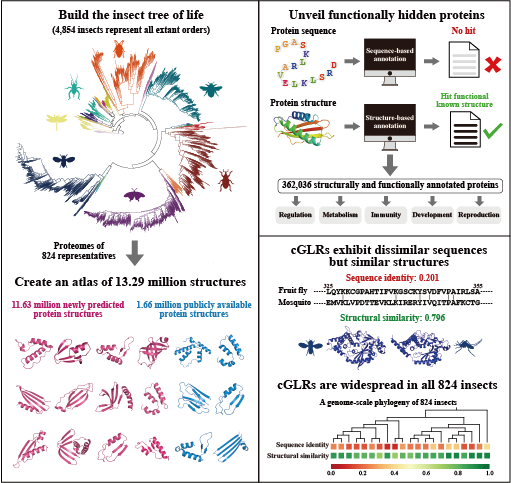
Protein structure connects sequence and function, allowing for in-depth exploration of biological processes across various organisms. Insects serve as an exceptional model for studying sequence, structure, and function relationships. We reconstructed a comprehensive phylogeny of 4,854 insects, representing all orders. Using this framework, we developed an atlas of 13.29 million predicted protein structures from 824 species, including 11.63 million newly predicted structures. Structural clustering indicated that proteins with dissimilar sequences but similar structures can be grouped together. We also identified 750 million remote homologs among insect proteins, many tracing back to ancient insect lineages. Notably, cGAS-like receptors (cGLRs) were structurally conserved across all 824 insects despite significant sequence divergence. Experimental assays confirmed that these cGLRs mediate innate immunity against virus infections in the yellow fever mosquito. Protein structures and their functions are available on the TIPS database.
◆9/20/2025 - Welcome Shixing, Sixuan, Zixin, Yujun and Yazhou to the Shen lab!
◆9/10/2024 - Welcome Qiancheng and Shuqin to the Shen lab!
◆6/4/2024 - Please check out our cover article in Nature Aging on discovery of a new longevity gene in animals!

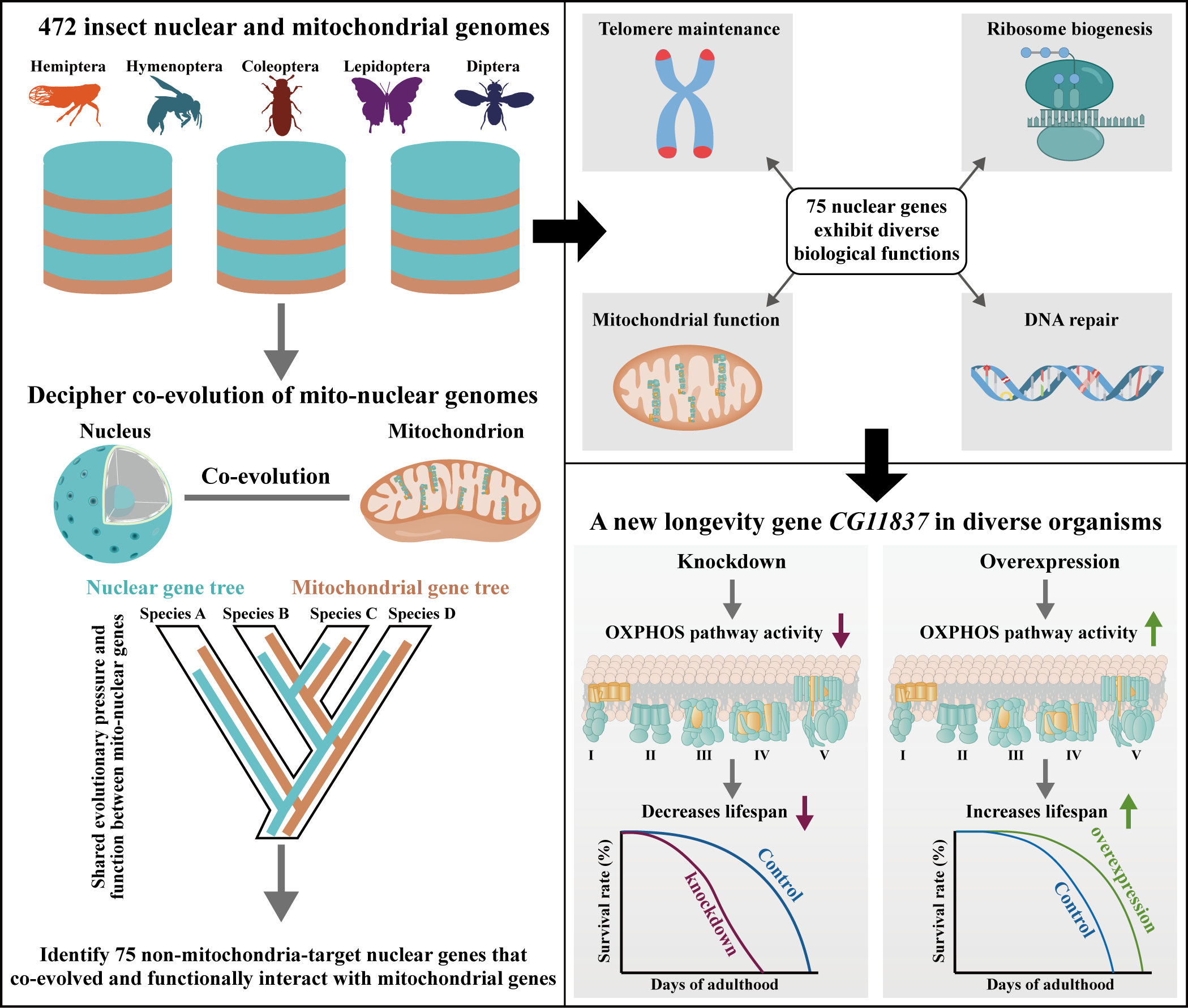
Oxidative phosphorylation (OXPHOS) is crucial for cellular energy synthesis, involving 13 mitochondrial and ~70 nuclear genes. While the evolutionary rate covariation of these genes is well-studied, the role of non-mitochondria-targeted nuclear genes remains unclear. By analyzing 13 mitochondrial genes and 1,417 nuclear BUSCO genes across 472 insects, we identified 75 nuclear genes with strong covariation with mtOXPHOS genes. Notably, the uncharacterized gene CG11837 was found to impact lifespan. CG11837 knockdown shortened lifespan in five insects and one worm, while overexpression extended lifespan in fruit flies and worms. Its human ortholog also protected cells from senescence. CG11837 enhances OXPHOS pathway activity, suggesting its role as a potential longevity gene across animals.
◆9/19/2023 - Welcome Xianfeng, Xinyi, Jiarong, and Jingxuan to the Shen lab!
◆12/22/2022 - The Shen lab was funded by the National Key Research and Development Young Program (No. 2022YFD1401600) and the National Science Foundation for Distinguished Young Scholars of Zhejiang Province (No. LR23C140001) !
◆12/14/2022 - Please check out new paper in Environmental Microbiology on open questions for HGTs in insects!
◆9/12/2022 - Welcome Weiyin and Jianchao!
We are very excited that two new members, Weiyin Wu and Jianchao Zhao, have joined the Shen lab! Weiyin will study phylogenomics. Jianchao will work on HGTs in animals.
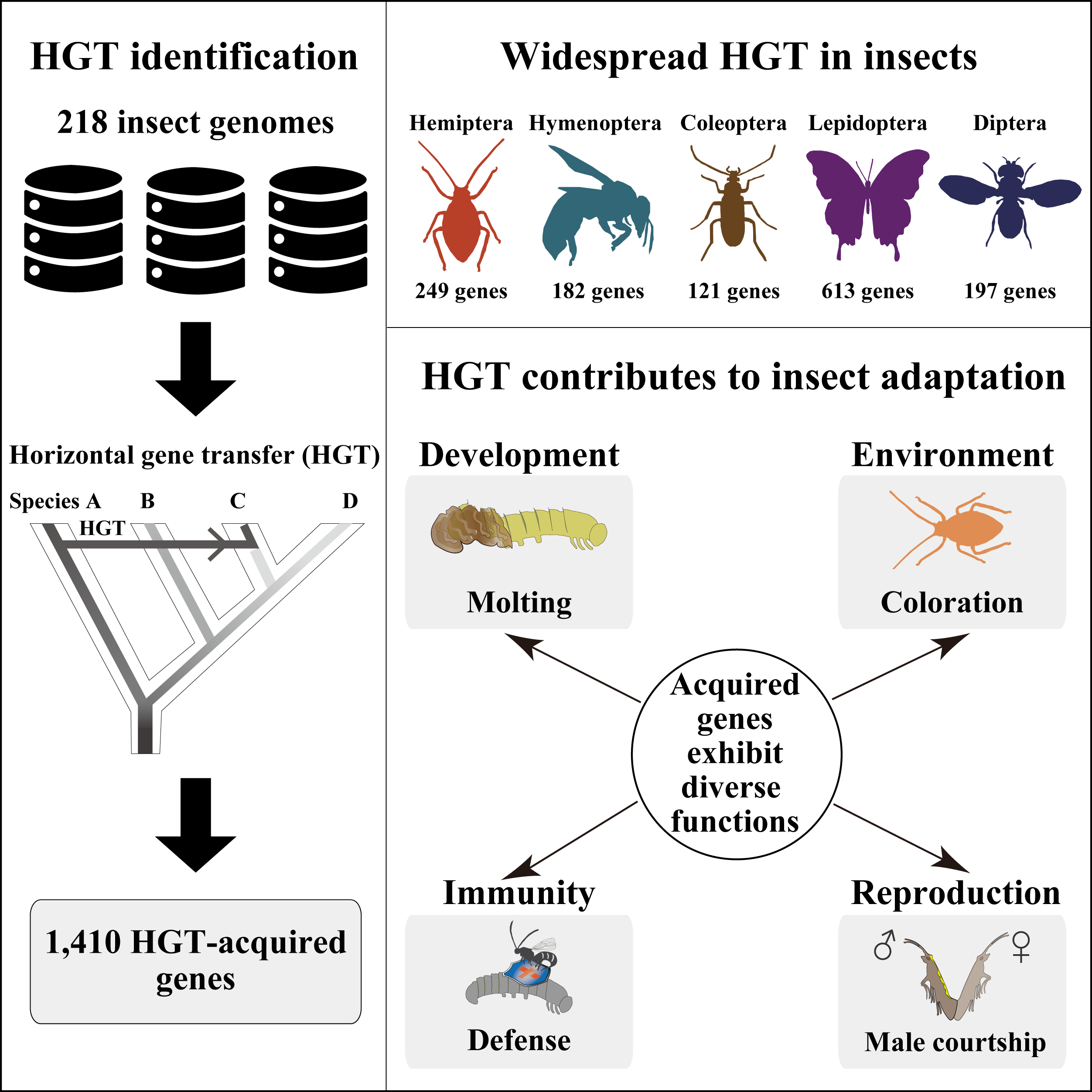
◆7/18/2022 - Please check out new paper in Cell on widespread horizontal gene transfer (HGT) in insects!

A foreign gene in moths and butterflies enhances
male courtship behavior
Mating experiments between wild-type and knockout
diamondback moths
In this study, we systematically examined horizontal gene transfers (HGTs) in 218 high-quality genomes of diverse insects and found that Lepidopterans (moths and butterflies) had the highest average number of HGT-acquired genes (16 genes per species). HGT-acquired genes containing introns exhibited substantially higher expression levels than genes lacking introns, suggesting that intron gains were likely involved in HGT adaptation. In addition, we used the CRISPR-Cas9 system to edit the most prevalent unreported gene LOC105383139, which was transferred into the last common ancestor of moths and butterflies. In diamondback moths, males lacking LOC105383139 courted females significantly less.
◆8/5/2021 - Welcome Yang, Yixiao, and Guozheng!
We are very excited that three new members, Yang Li, Yixiao Zhu, and Guozheng OU, have joined the Shen lab! Yang will focus on the evolution of foreign genes in animals. Yixiao will work on machine learning in phylogenetic inference. Guozheng will work on comparative mito-nuclear genomics across the eukaryotic tree of life.
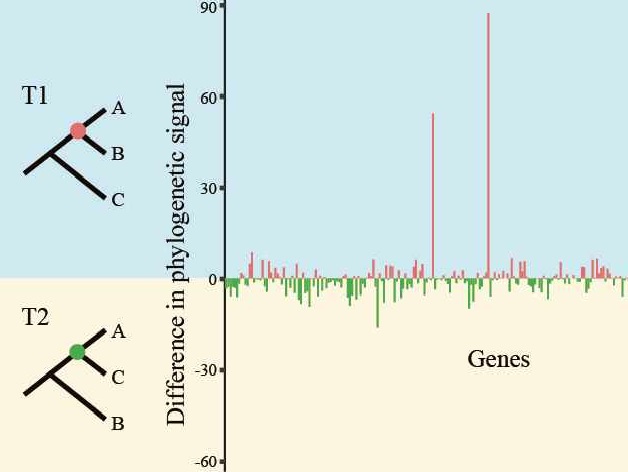
◆2/24/2021 - Please check out new paper in Systematic Biology on dissecting incongruence in phylogenomics!
We developed a workflow that can a) quantify the distribution of likelihood-based signal (measured by the difference in gene-wise log likelihood score or ΔGLS) and quartet-based topological signal (measured by the difference in gene-wise quartet score or ΔGQS) for every gene in a phylogenomic data matrix, and b) identify genes that exhibit inconsistent support between concatenation- and quartet-based approaches. Our results showed that removal of inconsistent genes eliminated or extensively reduced incongruence between concatenation- and quartet-based approaches. However, it should be noted that removal of inconsistent genes in simulated datasets reduced incongruence but did not always recover the true species phylogeny.
◆2/19/2021 - Please check out new paper published in Current Biology on fungal tree of life!
In this study, we explored the genome-scale phylogeny of the fungal kingdom based on 290 genes and 1,644 species and found ~85% of inferred phylogenetic relationships among fungi are robustly supported across different subsampling matrices and tree inferences. We found several unresolved relationships (e.g., Basidiomycota) may be due to ancient diversification events. Last, we found fungal higher rank taxonomy broadly reflects organisms' genome sequence divergence, with the exception of Saccharomycotina that current taxonomic rank assignments appear to not truly reflect their evolutionary divergence.
◆11/30/2020 - Please check out our new phylogenomic paper published in Nature Communications on irreproducibility in molecular phylogenetics!


In this study, we found that two replicate runs, that use exactly the same parameters (including substitution model, random starting seed number, tree search number, and log-likelihood epsilon value) on the same program (IQ-TREE or RAxML-NG), can generate substantially different maximum likelihood tree topologies. Interestingly, using 3 or more threads contributes to 2-fold increase in irreproducibility in IQ-TREE but not in RAxML-NG. Increasing reproducibility in ML inference will benefit from providing analyses' log files, which contain typically reported parameters (e.g., program, model, no. of tree searches) but also typically unreported ones (e.g., random starting seed, no. of threads, processor).
◆11/05/2020 - Our new paper just appeared in Science Advances on phylogenomics and contrasting modes of genome evolution in Ascomycota, the largest fungal phylum!
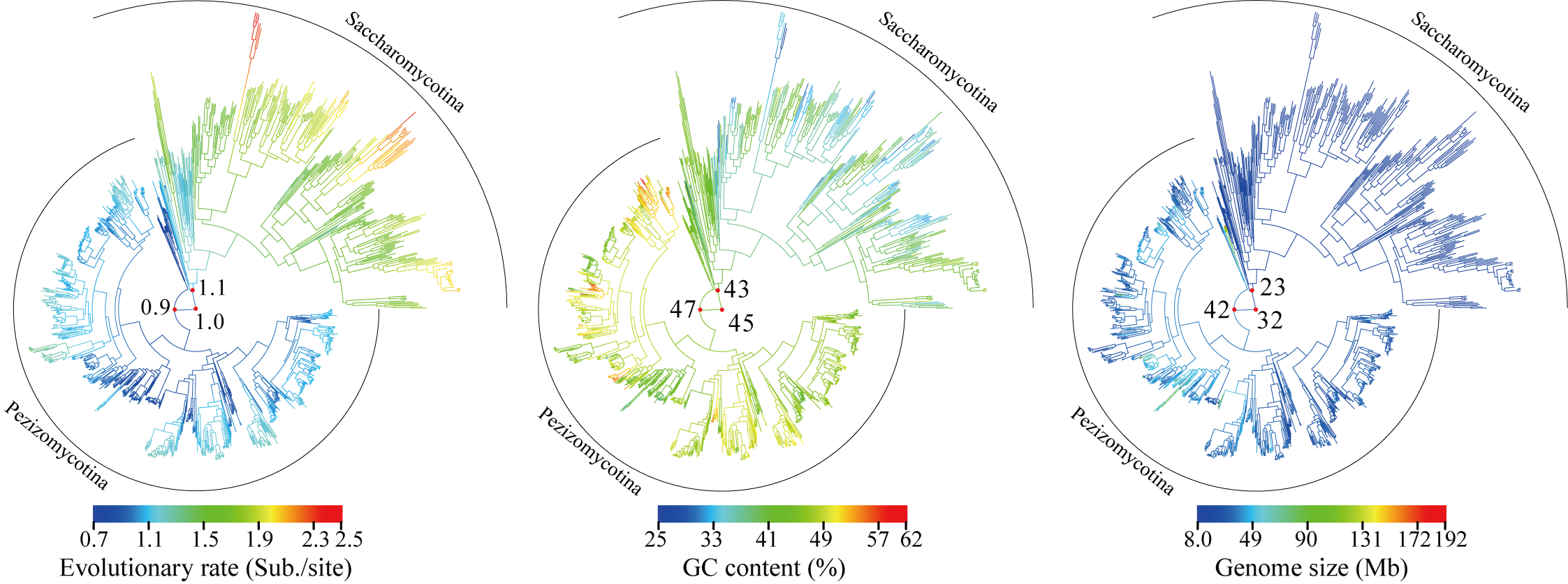
In this study, we provided a comprehensive phylogeny of the Ascomycota with 1,107 publicly available genomes (Saccharomycotina: 332; Pezizomycotina: 761; Taphrinomycotina: 14) and revealed that Saccharomycotina, which contains the single currently described class Saccharomycetes, and Pezizomycotina, which contains 16 classes, exhibited greatly contrasting evolutionary modes for seven genomic properties, in particular for evolutionary rate, GC content, and genome size. Our results provide a robust evolutionary framework for understanding the fundamental genetic and ecological processes that have generated the biodiversity of the largest fungal phylum.
◆09/19/2020 - The Shen lab was funded by the NSFC grant (No. 32071665)!
◆04/22/2019 - Xing-Xing Shen, 2019 Vanderbilt Postdoc of the Year Award, Honorable Mention!

